Amplicon sequencing reimagined
Amplicon sequencing is central to applications such as targeted and viral genotyping, characterizing genome edits, construct verification, and library diversity analysis. Traditional approaches like Sanger or short-read NGS can slow progress with complex workflows, limited read lengths, and long turnaround times.
Plasmidsaurus delivers a faster, more scalable alternative. Sequence amplicons from 100 bp to 25 kb using the latest Oxford Nanopore Technologies platform. With no need for sequencing primers, library prep, or cleanup, generate high-quality data and go from experiment to insight in a matter of hours.
Hassle-free setup
Sequence any sized amplicon with easy ordering. No sequencing primers, library prep, or purification required.
Accelerated science
Get your answers fast with linear/PCR results returned overnight and premium PCR and genotyping analysis results returned in 1-2 days.
Streamlined insights
Automated reporting answers your most critical questions.
Analyzing a mixture of variants? Use our new service Genotyping Analysis!
Characterize allelic composition and frequency at a single locus. 1-2 days turnaround, starting at $30.

Three services to meet your needs
As fast as
Next day
As low as
$15
Ideal for
Sequencing monoclonal PCR products
Typical applications
Determining or verifying the sequence of a DNA construct or genomic region
Whole viral genome sequencing
Data deliverables
One consensus sequence
Raw reads from your sample
As fast as
1 - 2 days
As low as
$30
Ideal for
Sequencing PCR products when working with mixed populations of molecules, or when sufficient read depth or full length reads are required
Typical applications
Characterizing CRISPR editing efficiency
Quantifying and assessing library diversity
Data deliverables
One consensus sequence
Raw reads from your sample
As fast as
1 - 2 days
As low as
$30
Ideal for
Phasing variants across a wide range of allele frequencies
Typical applications
Cell line, model organism, or germplasm QC
Locus-specific genotyping
Characterizing genetic crosses
Biomarker-based stratification
Data deliverables
Visualizations comparing aligned alleles, frequencies of alleles and fasta sequences of each predicted allele above 10%
Raw reads from your sample
Key applications
Cloning
Confirm correct sequences at every step of your workflow.
Gene editing screening and confirmation
Characterize editing system performance and screen for cells with the right edit.
Genotyping
Verify or determine the genotype of your sample in the target region. Learn more about our Genotyping Analysis service here.
Viral genome sequencing
Accurate whole genome sequencing for viral evolution, epidemiological research, and pathogen surveillance.
Characterize library diversity
Characterize the complexity and diversity of any linear-DNA based library.
"And thank you@plasmidsaurus for their quick PCR sequencing service that put me on the path to understanding. You guys provide a rock solid service. IMO you are to sequencing what NEB is to enzymes."
Level up your amplicon sequencing
Let us do the cleanup for you
PCR cleanup takes time better spent on real science. Add on cleanup for $5 and send us samples straight from the PCR machine—we’ll handle the rest. Get the same great results with less fuss.
Data deliverables & bioinformatics
For visualizations comparing aligned alleles, allele frequencies, and sequences, learn more about Genotyping Analysis here.
Virtual gel
Consensus sequence
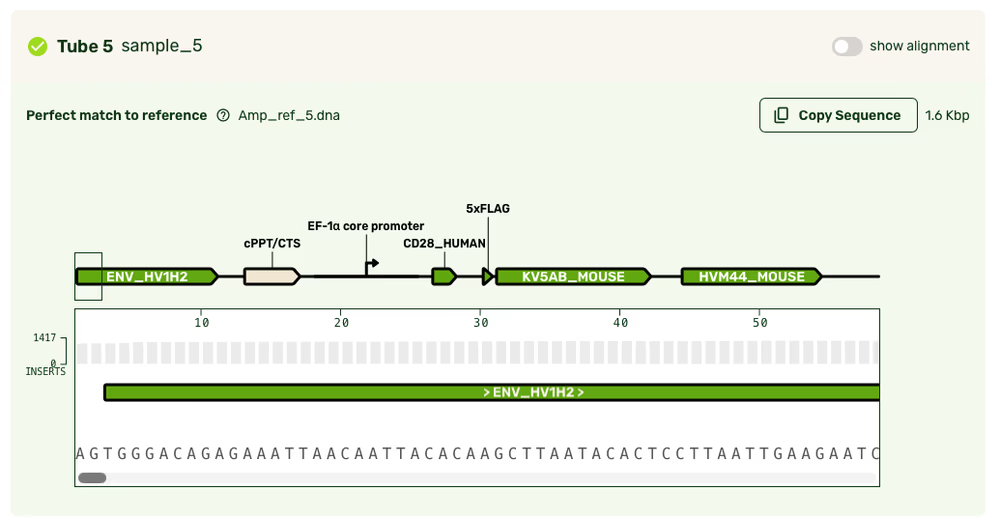
Feature annotations
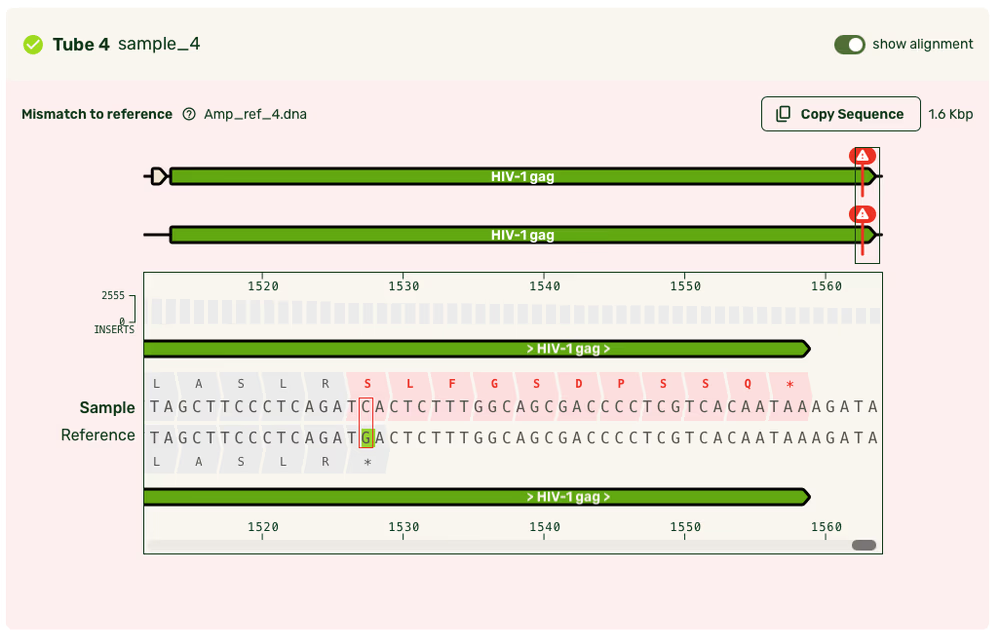
Reference alignment
Sequencing statistics including read length and coverage
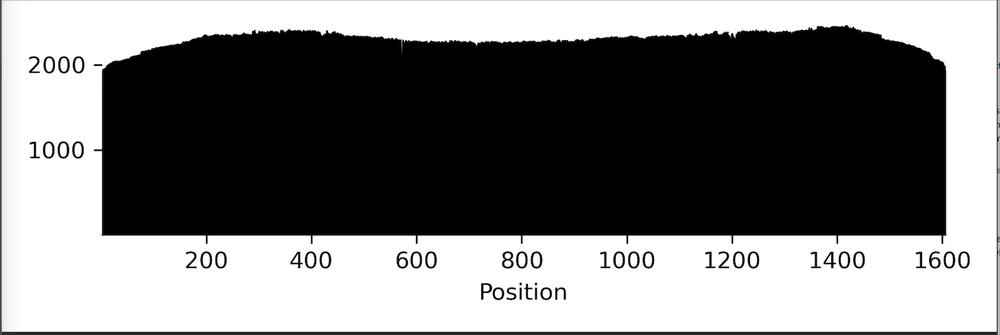
Raw reads
For Premium PCR, note that for complex mixtures where it is not possible to generate a consensus, only a virtual gel, read length histogram, and raw reads with annotations are returned.
Product specs & service levels
- Long read sequencing with ONT
- Optimized for clonal, linear, double-stranded DNA
| Service Level | Size | Minimum Volume | Concentration | Cost | Target Turnaround Time |
|---|---|---|---|---|---|
| Standard unpurified | 500 bp - 25 kb | 10 μL | 20 - 200 ng/μL | $15 | 1 day |
| Standard unpurified + optional cleanup | 500 bp - 25 kb | 10 μL | 20 - 200 ng/μL | $20 | |
| Standard purified | 500 bp - 25 kb | 10 μL | 20 - 200 ng/μL | $15 | |
| Big purified | 25 - 125 kb | 20 μL | 50 - 400 ng/μL | $30 |
- Long read sequencing with ONT
- Optimized for linear, double-stranded DNA
- Add sample cleanup for $5
| Service Level | Size | Target Read Depth/Sample* | Minimum Volume | Concentration** | Cost | Target Turnaround Time |
|---|---|---|---|---|---|---|
| Standard | 100 bp - 25 kb | Up to 3,000 reads | 10 μL | 1 ng/μL per 100 bp | $30 | 2 days |
| Big | 100 bp - 25 kb | Up to 6,000 reads | 20 μL | 1 ng/μL per 100 bp | $60 | |
| Huge | 100 bp - 25 kb | Up to 12,000 reads | 40 μL | 1 ng/μL per 100 bp | $120 |
* Greater than 12,000 reads per sample can be accommodated through our Custom Sequencing Service.
** Sequencing is based on the molarity of your DNA insert, therefore the required concentration (ng/uL) will vary depending on your insert size.
- Long read sequencing with ONT
- Optimized for linear, double-stranded DNA
- Add sample cleanup for $5
| Service Level | Size | Target Read Depth/Sample* | Minimum Volume | Concentration* | Cost | Target Turnaround Time |
|---|---|---|---|---|---|---|
| Standard | 100 bp - 25 kb | Up to 3,000 reads | 10 μL | 1 ng/μL per 100 bp | $30 | 1-2 days |
| Big | 100 bp - 25 kb | Up to 6,000 reads | 20 μL | 1 ng/μL per 100 bp | $60 | |
| Huge | 100 bp - 25 kb | Up to 12,000 reads | 40 μL | 1 ng/μL per 100 bp | $120 |
* Sequencing is based on the molarity of your DNA insert, therefore the required concentration (ng/uL) will vary depending on your insert size.
Ready to sequence?
Put your DNA in a tube and drop it off. Click below for requirements and suggestion for optimal results.
Relevant resources



Related products
Microbiome
Exploring microbial diversity? Check out our Microbiome Sequencing to receive species-level classification for your 16S, 18S and/or ITS full length gene amplicons.
AAV
Sequencing AAV? Check out our AAV sequencing service for an ultra-fast service that returns comprehensive data with minimal sample input.
Whole Plasmid
Our Whole Plasmid Sequencing service with ZeroPrep lets you skip culture and minipreps and get straight to your results faster.
FAQs
We sequence each sample with Oxford Nanopore long reads to very high depth before generating a consensus/assembly using the latest basecalling and polishing software:
- We construct an amplification-free long-read sequencing library using the newest v14 library prep chemistry, including minimal fragmentation of the input linear DNA in a sequence-independent manner
- We sequence the library with a primer-free protocol using the most accurate R10.4.1 flow cells (raw data is delivered in .fastq format).
- We use the re-assembled raw reads to generate a high-accuracy linear consensus sequence from the raw reads.
- For standard linear/PCR samples, we will also return a set of feature annotations.
We use the latest flowcells and chemistry kits from Oxford Nanopore, along with the latest Super Accurate basecalling model. Amplicon consensus accuracy is often above Q60, which corresponds to 99.9999%, or one error per 1,000,000 bases.
Results for the Linear/PCR service are typically returned next business day, and results for Premium PCR service are typically returned within 1-2 business days.
This is the average time it takes to return data to customers who are submitting orders from US, UK and EU. Turnaround times for customers in APAC may be slightly longer due to the time required in transit to our lab in Singapore.
Low sample quality can also negatively impact turnaround time and the quality of your results. Please read sample prep instructions carefully to make sure your samples meet input requirements. Due to variability in shipping logistics and sample quality that is outside our control we cannot guarantee turnaround times or sample success rates.
Our Linear/PCR product works in the same way as our Whole Plasmid service: you will receive an annotated assembled consensus of your submitted amplicon. The exact number of reads used to create this assembly varies based our internal quality control metrics, but is typically over a thousand.
The amount of sequencing coverage you will receive for Premium PCR is dependent on which option you choose:
| Category | Amplicon Size | Read Depth | Minimum Volume | Concentration | Cost (USD) |
| Standard | 100 bp - 25 kb | Up to 3k reads | 10 μL | 1 ng/μL per 100 bp | $30 |
| Big | 100 bp - 25 kb | Up to 6k reads | 20 μL | 1 ng/μL per 100 bp | $60 |
| Huge | 100 bp - 25 kb | Up to 12k reads | 40 μL | 1 ng/μL per 100 bp | $120 |