Rethink your microbiome workflow
Most microbiome studies still depend on large batched sequencing runs that take weeks and only resolve to the genus level. Plasmidsaurus Microbiome Sequencing offers more agile options to accelerate your research:
- Full-length 16S, 18S, or ITS reads with species-level classification, delivered as quickly as overnight
- Comprehensive shotgun metagenomics (now in beta) for a complete genomic picture of your microbial community
Designed for speed, precision, and flexibility, Plasmidsaurus microbiome sequencing services are ideal for uncovering microbial drivers of human health and disease, monitoring environmental communities, and understanding how experimental biomes respond to stimuli. No matter your research question, we provide the high-resolution data you need, fast.
Confirm what's in your community
Confirm your system has the expected species diversity before starting your experiment, with insights direct from raw samples, genomic DNA, or PCR amplicons.
Monitor with rapid results
Easily monitor your microbial communities over time, across locations, and in response to stimuli, with taxonomic classification as fast as overnight.
Discover with species-level clarity
Precisely identify microbial species that traditional microbiome sequencing sequencing can’t resolve. Discover what species are present and in what relative abundances.
Which service is right for me?
Sample input
Extracted environmental DNA OR raw samples such as soil, feces or swabs (within US only)
Ideal for
Exploring the bacteria and archaea composition of samples with high-host (e.g. human) DNA
Service levels
Standard: Up to 5,000 reads
Big: Up to 10,000 reads
Huge: Up to 20,000 reads
Bronto: Up to 500,000 reads
Sequencing configuration
Oxford Nanopore long reads
As fast as
1 - 3 days
As low as
$45
Sample input
Purified amplicons of full-length 16S, 18S or ITS genes, or a mixture thereof
Ideal for
Studying fungi, protists, algae or other microbes that lack 16S genes
Service levels
Standard: Up to 5,000 reads
Big: Up to 10,000 reads
Huge: Up to 20,000 reads
Add cleanup for $5
Sequencing configuration
Oxford Nanopore long reads
As fast as
Next day
As low as
$30
Sample input
Extracted environmental DNA
Ideal for
Comprehensively profiling of diverse microbial communities across all kingdoms
Service levels
Standard: 2 Gb (6.7M read pairs)
Big: 10 Gb (33.3M read pairs)
Sequencing configuration
Illumina 2 x 150 paired-end reads
As fast as
6-8 days
As low as
$90
New! Shotgun Metagenomic Sequencing now available in Beta
Our new service for comprehensively profiling complex communities. Trace your microbial community from domain to species with interactive taxonomy exploration.
Product specs & service levels
All samples are sequenced with ONT.
| Service Level | Sample Input | Read Depth/Sample | Minimum Volume | Concentration | Cost | Target Turnaround Time |
|---|---|---|---|---|---|---|
| Standard | gDNA | Up to 5,000 | 10 µL min | 10 ng/µL | $45 | 1 business day |
| Big | Up to 10,000 | 20 µL min | $75 | |||
| Huge | Up to 20,000 | 40 µL min | $135 | |||
| Bronto | Up to 500,000 | 40 µL min | $250 |
Add DNA extraction from raw samples for $15 extra and 3 business day turnaround time.
All samples are sequenced with ONT. Add cleanup for $5 while placing your order. Cleanup adds 2 days to expected turnaround times.
| Service Level | Sample Input | Read Depth/Sample | Minimum Volume | Concentration | Cost | Target Turnaround Time |
|---|---|---|---|---|---|---|
| Standard | Linear double-stranded DNA | Up to 5,000 | 10 µL min | 10-20 ng/µL | $30 | 1 business day |
| Big | Up to 10,000 | 20 µL min | $60 | |||
| Huge | Up to 20,000 | 40 µL min | $120 |
All samples are sequenced with Illumina 2 x 150 paired-end configuration.
| Service Level | Sample Input | Raw Data/Sample* | Cost | Target Turnaround Time |
|---|---|---|---|---|
| Standard | gDNA | 2 Gb (6.7M read pairs) | $90 | 6-8 business days |
| Big | 10 Gb (33.3M read pairs) | $140 |
* Please note that because this untargeted approach sequences all DNA in your sample, host DNA (e.g., human) will take up a portion of the 2 Gb or 10 Gb of sequencing depth
Data deliverables & bioinformatics
Taxonomic diversity
Read identifications
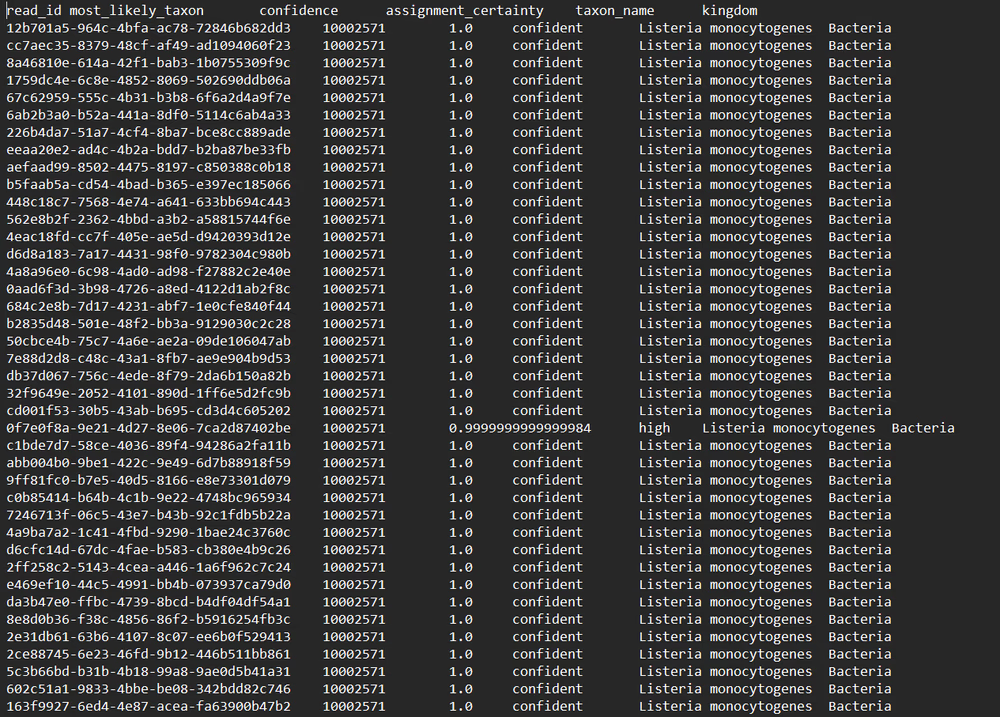
[Microbiome 16S Amplification & Sequencing and Microbiome Amplicon Only]
Summary
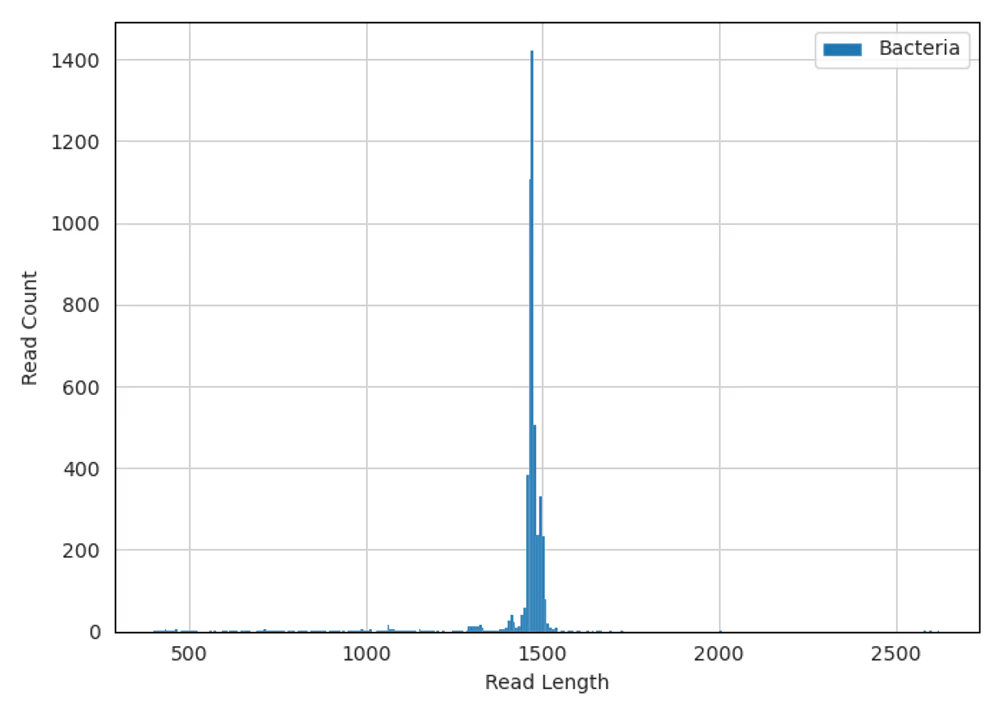
Raw reads
Ready to sequence?
Send your messy sample, purified DNA, or PCR amplicon. We'll handle the rest.
Relevant resources

Related products
Whole Genome
Want a deeper look at one organism in your community? Try Whole Genome Sequencing for a complete, assembled, annotated sequence of your clonal isolate.
Amplicon
Monitoring a different gene? Try Amplicon Sequencing instead.
Custom
Have a unique sequencing case? Work with us on custom sequencing.
FAQs
We use in-house gene amplification or PCR primers to perform barcoded full-length amplification of the 16S gene and construct an Oxford Nanopore long-read sequencing library using the newest v14 library prep chemistry. We sequence the amplicons with primer-free protocol using the most accurate R10.4.1 flow cells producing a full-length sequencing read for each amplicon.
We perform end-ligation of your 16S, 18S and/or ITS amplicons and construct an Oxford Nanopore long-read sequencing library using the newest v14 library prep chemistry. We sequence the amplicons with primer-free protocol using the most accurate R10.4.1 flow cells, producing a full-length sequencing read for each amplicon.
Plasmidsaurus Shotgun Metagenomics is performed using the newest short-read sequencing technology from Illumina. We use transposase-mediated library preparation chemistry to profile the total genomic DNA present in your sample—capturing bacteria, archaea, fungi, and viruses in a single assay—and return a comprehensive taxonomic analysis of your microbial community.
There are numerous approaches to extracting metagenomic DNA from the source material (swabs, soil, feces, et cetera), so we do not provide specific recommendations. Any extraction method that yields high quality, high purity, high molecular weight, double-stranded gDNA that is free of nicks, gaps, breaks, and contaminants (enzymatic inhibitors, RNA, etc.) is suitable for this service.
See sample prep for more information.
Zymo DNA/RNA Shield™-preserved source materials for extraction cannot be shipped via your local US dropbox, but instead must be shipped directly to our Eugene lab. You can request a free shipping label during order submission or ship on your own to:
Plasmidsaurus
1850 Millrace Drive, Suite 200
Eugene, OR 97403