Never use outdated sequencing tools again
We pioneered whole plasmid sequencing to solve a problem that had plagued biologists for decades. For too long, researchers were stuck with 1970s sequencing methods that revealed only fragments of their DNA constructs, leading to invisible backbone errors, painful project failures, and weeks of wasted troubleshooting. Nearly 5 million plasmids later, we’re still innovating to make sequencing work for scientists, helping you skip the headaches and get answers.
Quality you can depend on
Robust processes and our proprietary analysis pipeline ensure accurate data even with low concentration, low copy, or difficult plasmids.
Easy insights
Get high quality bioinformatics analysis straight to your inbox, highlighting mismatches, duplications, and other errors before they derail your science.
Overnight results, zero extra steps
No primers or minipreps needed. Cut out extra steps in sample prep and data analysis so your science can move faster than ever before.
Goodbye minipreps, hello answers
Tired of miniprepping? Send us an agar plate, liquid culture, or resuspended colony and go straight from colony to whole plasmid sequence overnight for an extra $5.
Join Over 70,000 Scientists
Accelerate your research with Plasmidsaurus. What would you do with more time?
Put some plasmids in the dropbox at 3pm yesterday and got the results at 02:55am. Less than 12 hours for the full annotated sequence of 4 plasmids is quite mind boggling.
If it weren’t for Plasmidsaurus, we’d easily be 6–9 months behind where we are now. Short design-build-test-learn cycles are absolutely key.
Without Plasmidsaurus we couldn’t confidently sequence our constructs. It has been a game changer for us, saving us both time and money.
The most significant improvement Plasmidsaurus provides over competitors is their failure rate was pretty high while Plasmidsaurus’ approached zero.
We’ve been sequencing with Plasmidsaurus to validate clones. It’s faster and more comprehensive than ordering cohorts of Sanger primers.
Shoutout to @Plasmidsaurus for not only sequencing my 12 samples in less than a day but also being able to identify stuff like entire plasmid duplications!
Plasmidsaurus identified backbone errors in plasmids cloned by our gene synthesis company, which we could never have identified via Sanger.
Why choose Plasmidsaurus?
Highest quality
Recognized by Oxford Nanopore as the Gold Standard, our custom analysis pipelines produce the highest quality data with the fewest sequencing errors.
Most robust workflow
Our optimized processes ensure that >99.9% of samples that meet minimum quality thresholds are sequenced successfully
Insights to your inbox
Only Plasmidsaurus offers automated analysis that answer your most critical questions (Is my sequence correct? Is my sample contaminated?) so you can skip data analysis and focus on your next experiment.
Sequencing made simple. No bioinformatician needed.
Get confidence in every base
See basecalls for each position across all sequencing reads.
Zero in on mutations, insertions, and deletions
Catch errors quickly with base-level insights and amino acid consequences.
Results in your inbox
View overnight results quickly before diving in for deeper analysis.
Try out our live demo with real data
See what Sanger is missing
Verify plasmid identity, purity, and stability.
Data deliverables & bioinformatics
Consensus sequence (.fasta, .gbk)
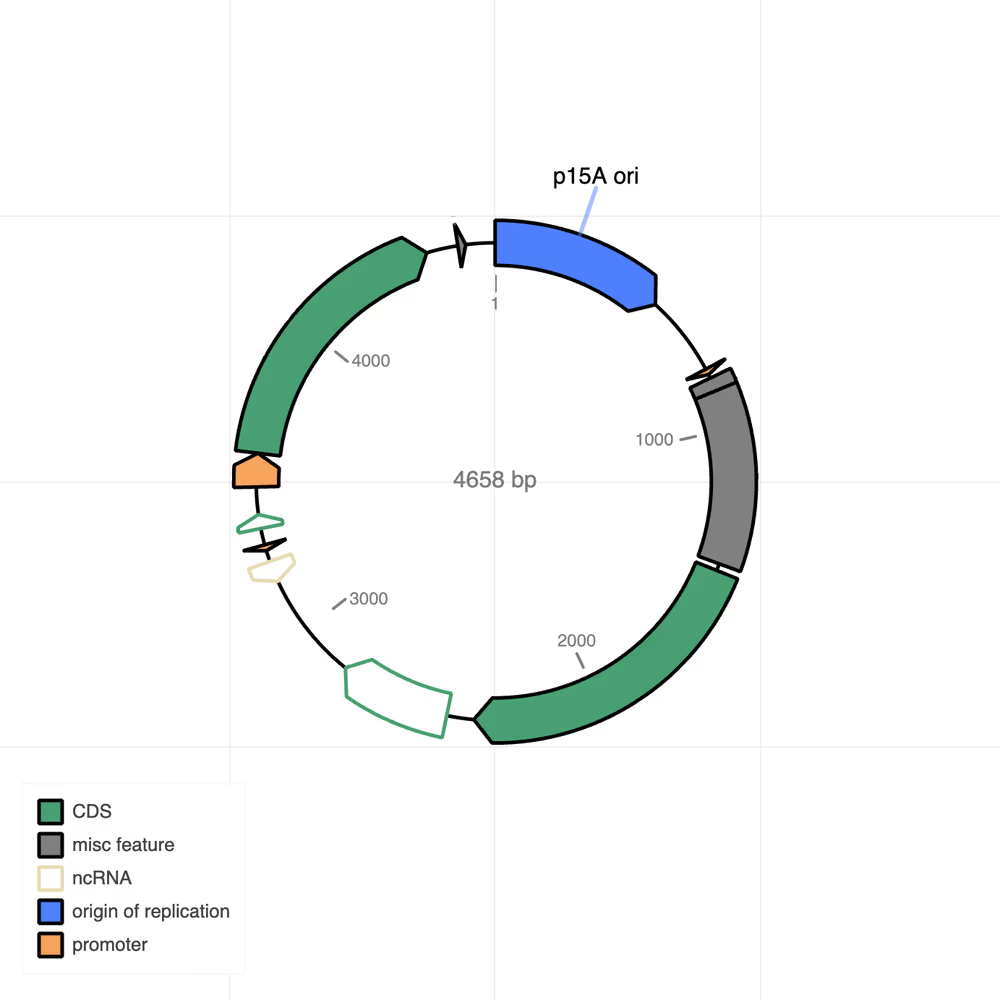
Plasmid map (.html)
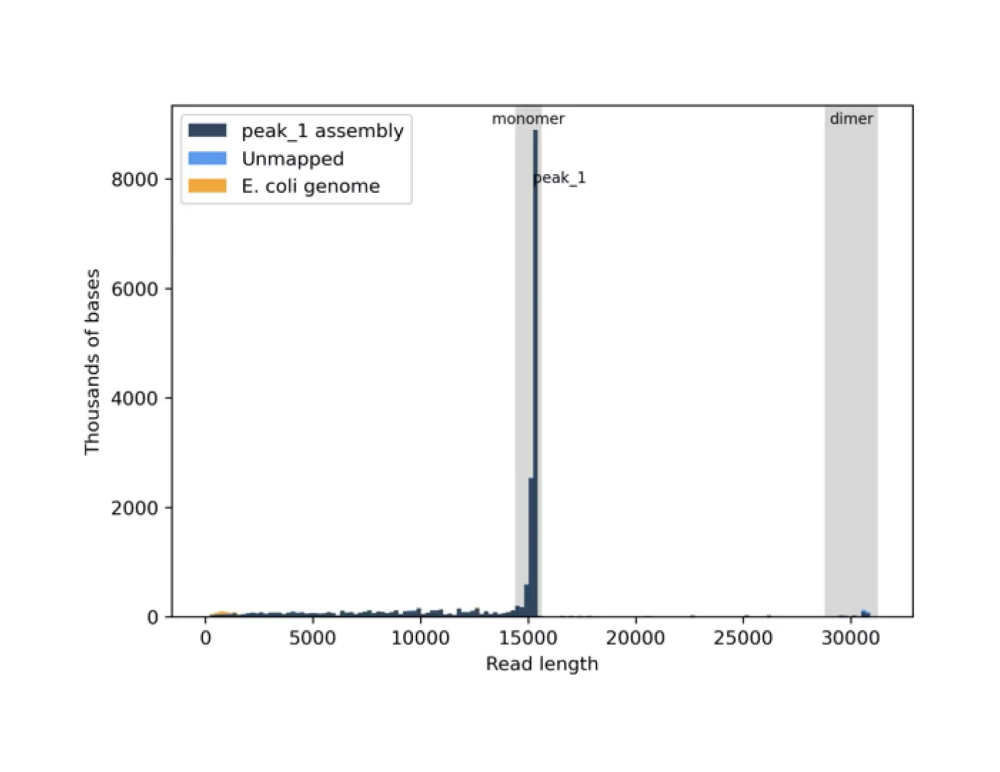
Read length histogram (.png)
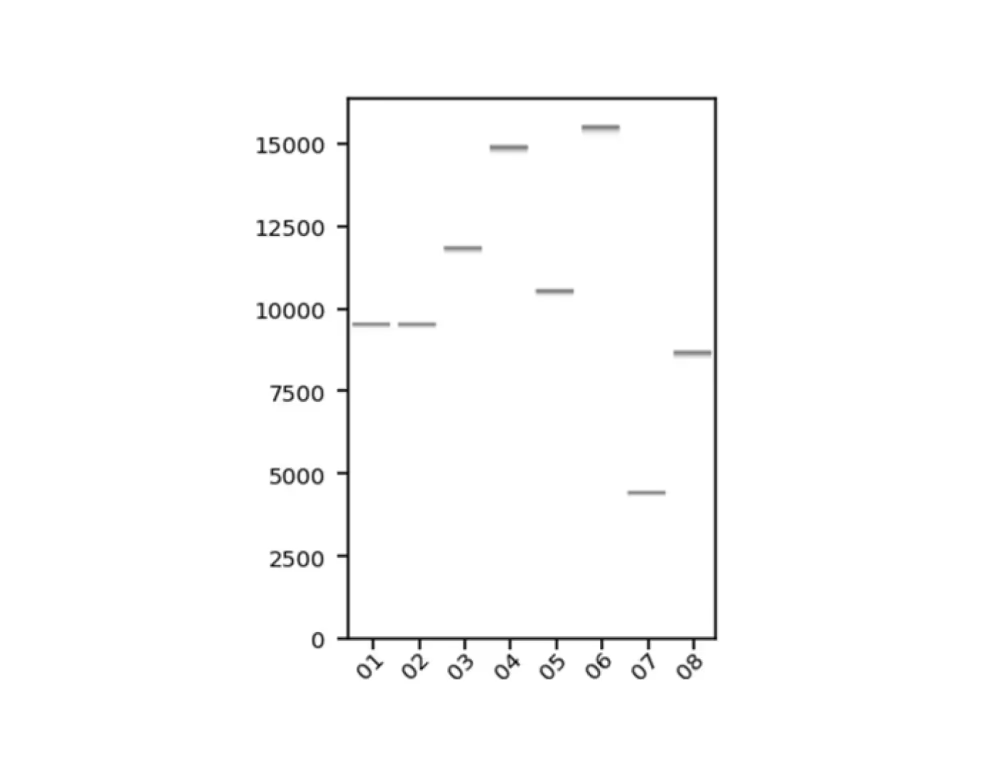
Virtual gel (.png)
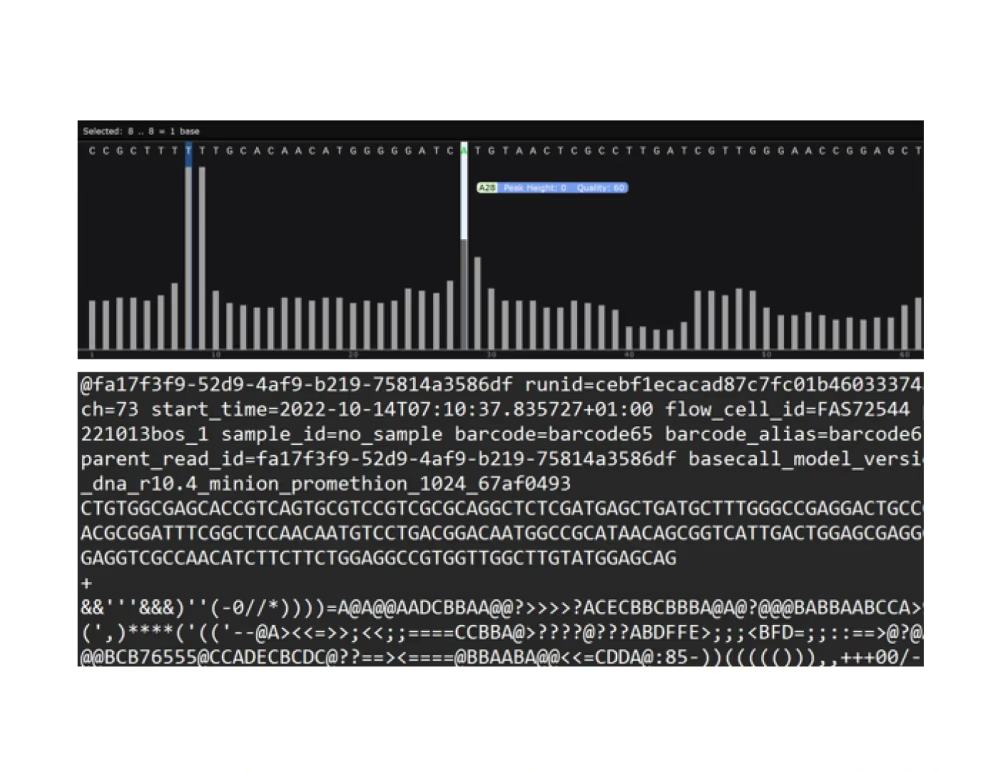
Chromatogram (.ab1, .gbk)

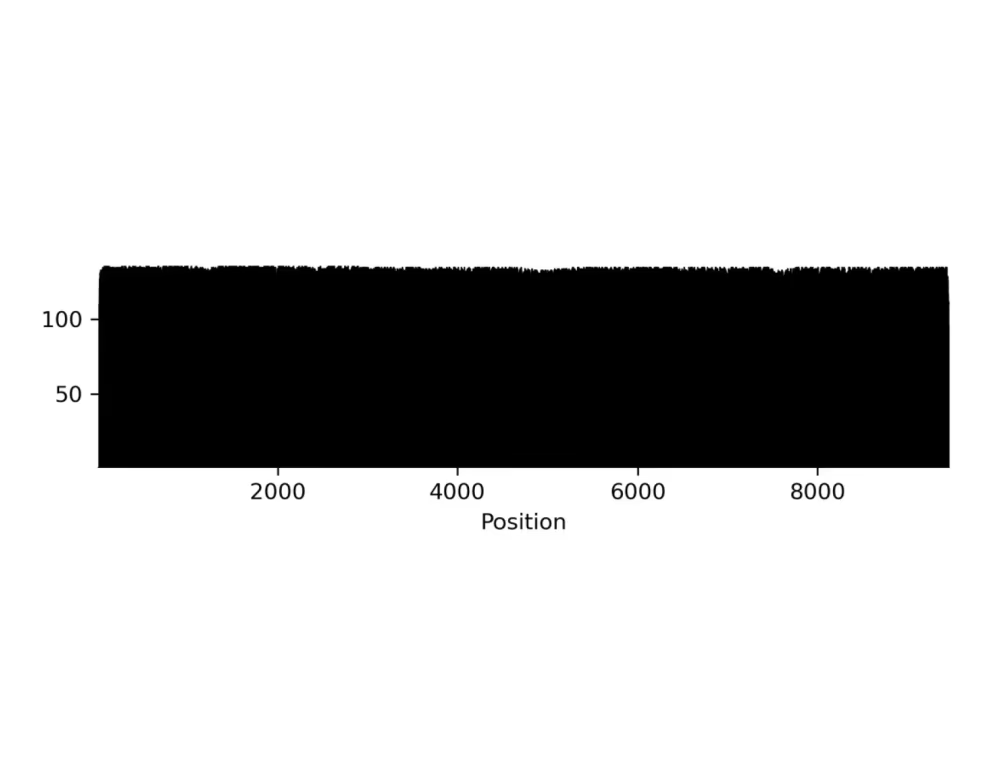
Coverage plot (.png, .gbk)
Per-base data (.txt, .tsv)
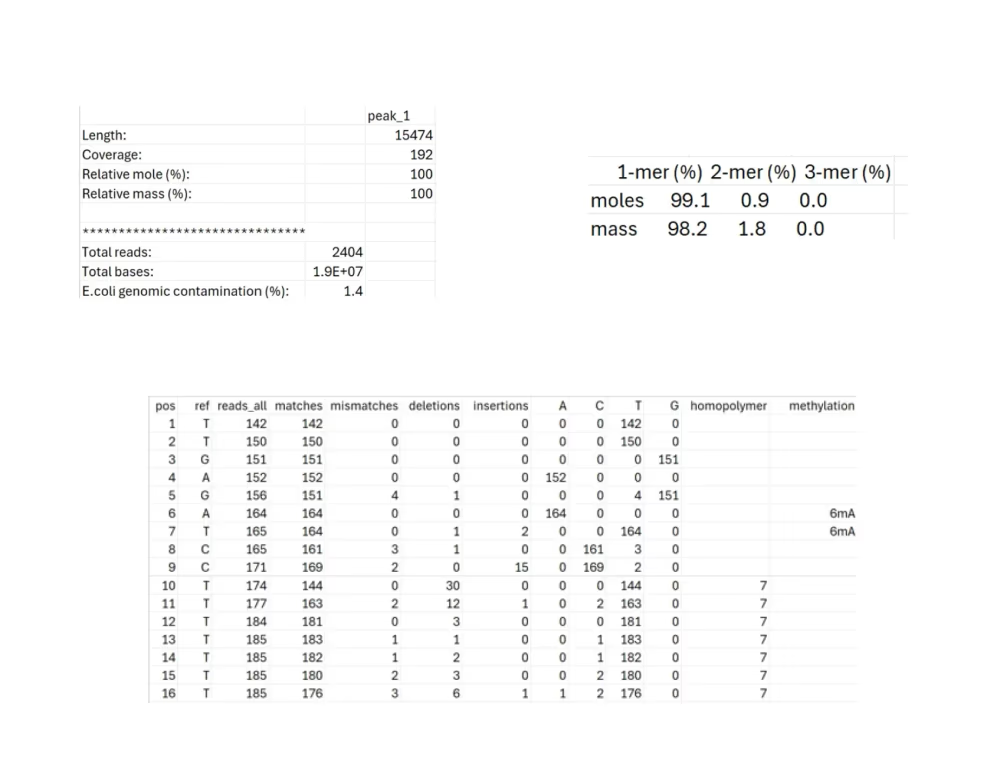
<ul>
<li>SAMPLE.tsv: Indicates how well the raw reads agree with the consensus sequence at each position. </li>
<li>SAMPLE_multimer_analysis.txt: Indicates the % distribution of the various concatemer forms of the consensus sequence (monomer, dimer, trimer, etc.)</li>
<li>SAMPLE_summary.tsv: Indicates the length, average coverage, relative composition (by moles and mass), total reads, total bases, and % E. coli genomic DNA contamination for the consensus sequence.</li>
</ul>
Raw read sequenced (.fastq.gz)
Product specs & service levels
| Service Level | Size | Concentration | Volume | Purity | Cost | Target Turnaround Time |
|---|---|---|---|---|---|---|
| Standard Low Concentration | <25 kb | 20 - 200 ng/µL | 10 µL | Column or bead purified | $15 | 1 day |
| Standard High Concentration | <25 kb | 200 - 1000 ng/µL | 4 µL | $15 | ||
| Big | 25 - 125 kb | 50 - 400 ng/µL | 20 µL | $30 | ||
| Huge | 125 - 300kb | 50 - 400 ng/µL | 40 µL | $60 |
| Service Level | Sample Input | Plasmid Size | Cost | Target Turnaround Time |
|---|---|---|---|---|
| Liquid Culture | 20 μL - 100 μL of turbid culture in LB media, OD600 > 0.3 (recommended) Liquid should appear cloudy upon submission | <20 kb | $20 | 1 day |
| Colony Resuspension | >2 mm colony resuspended in 20 μL of LB media (recommended) Liquid should appear cloudy upon submission | |||
| Agar Plate Colony | ~1 cm2 patches clearly labeled OR >2 mm colonies clearly circled and labeled |
Ready to sequence?
Send us extracted and purified plasmid DNA, or go straight from colony to sequence.
Relevant resources

Related products
Whole Genome
Want to sequence an entire genome? Use Whole Genome Sequencing for nearly any organism.
Amplicon
Need to verify cloning and gene editing with a specific region? Try Amplicon Sequencing.
Custom
Have sequences that are really small or really big? Have large plasmid variant libraries? Contact us via Custom Sequencing services for more options.
FAQs
Plasmidsaurus Whole Plasmid Sequencing is performed using the newest long-read sequencing chemistry from Oxford Nanopore Technologies (ONT).
We use a transposome complex to cut your circular plasmid at a random point, creating an amplification-free library of full-length linear molecules, so that each sequencing read spans the entirety of the plasmid. We return the full length consensus sequence of your plasmid, beginning at the origin.
We do not guarantee a specific level of coverage as the number of raw reads generated can vary due to length and sample quality, though typically samples yield hundreds of raw sequencing reads. These go through multiple quality filtering steps to ensure we are delivering the best possible assembly.
We use the latest flowcells and chemistry kits from Oxford Nanopore, along with the latest Super Accurate basecalling model. The vast majorities of our plasmid assemblies contain no errors compared to the reference, which you can read more about here. Consensus accuracy is often above Q60, which corresponds to 99.9999%, or one error per 1,000,000 bases.
We do not guarantee a specific level of coverage as the number of raw reads generated can vary due to length and sample quality, though typically samples yield thousands of raw sequencing reads. These go through multiple quality filtering steps to ensure we are delivering the best possible assembly.
Average coverage is reported in the SAMPLE_summary.tsv file, and coverage over ~20x indicates a very accurate consensus.
This service is intended for a clonal population of molecules. If your species are very similar (e.g. differ by only a few nucleotides), the pipeline will most likely create a single consensus file, with mixed peaks observed in the .ab1 file where there are SNPs and indels.
If your species are sufficiently distinct in size or sequence, the pipeline will generate a single consensus sequence for the molecular species that produces the largest amounts of total sequencing data. (Please note that multimer forms such as dimers, trimers, etc. are not considered different molecular species by the pipeline, so you will only receive the monomer consensus sequence by default).
Ultimately, which species ends up producing a consensus will vary depending on overall sample quality, coverage, and relative abundance of each species. Sequencing is considered successful if the pipeline is able to generate a consensus, even if it is not your target.
Re-sequencing mixtures won't change the relative proportions of the species (and hence which species generate a consensus), but you can submit multiple aliquots if you need higher overall coverage. Custom sequencing is available to sequence mixed populations (e.g. large barcode or variant libraries). You can submit requests for custom projects via the Custom sequencing page.