Resource Center
FAQ
Whole Plasmid
Technical Details
Plasmidsaurus Whole Plasmid Sequencing is performed using the newest long-read sequencing chemistry from Oxford Nanopore Technologies (ONT).
We use a transposome complex to cut your circular plasmid at a random point, creating an amplification-free library of full-length linear molecules, so that each sequencing read spans the entirety of the plasmid. We return the full length consensus sequence of your plasmid, beginning at the origin.
We do not guarantee a specific level of coverage as the number of raw reads generated can vary due to length and sample quality, though typically samples yield hundreds of raw sequencing reads. These go through multiple quality filtering steps to ensure we are delivering the best possible assembly.
We use the latest flowcells and chemistry kits from Oxford Nanopore, along with the latest Super Accurate basecalling model. The vast majorities of our plasmid assemblies contain no errors compared to the reference, which you can read more about here. Consensus accuracy is often above Q60, which corresponds to 99.9999%, or one error per 1,000,000 bases.
Lots of reasons! Here are a few:
- Scientific rigor and peace of mind
- E. coli and other hosts will go to great lengths to avoid expressing your leaky toxic gene, including modifying your plasmid in unexpected ways that are invisible to Sanger sequencing.
- Plasmid inserts are getting longer and more complex. Instead of multiple Sanger runs, or synthesizing a sequencing primer, or doing primer walking, simply sequence the whole plasmid in a single read
- Long reads are ideal for resolving repetitive regions that totally flummox Sanger sequencing
- Are you sure your plasmid isn't a dimer? Are you sure there aren't multiple plasmids in your strain? Sanger sequencing won't tell you, and we see it every single day
- For a similar price per sample you get significantly more information, and a QC test that can properly assess your plasmid
This service is intended for a clonal population of molecules. If your species are very similar (e.g. differ by only a few nucleotides), the pipeline will most likely create a single consensus file, with mixed peaks observed in the .ab1 file where there are SNPs and indels.
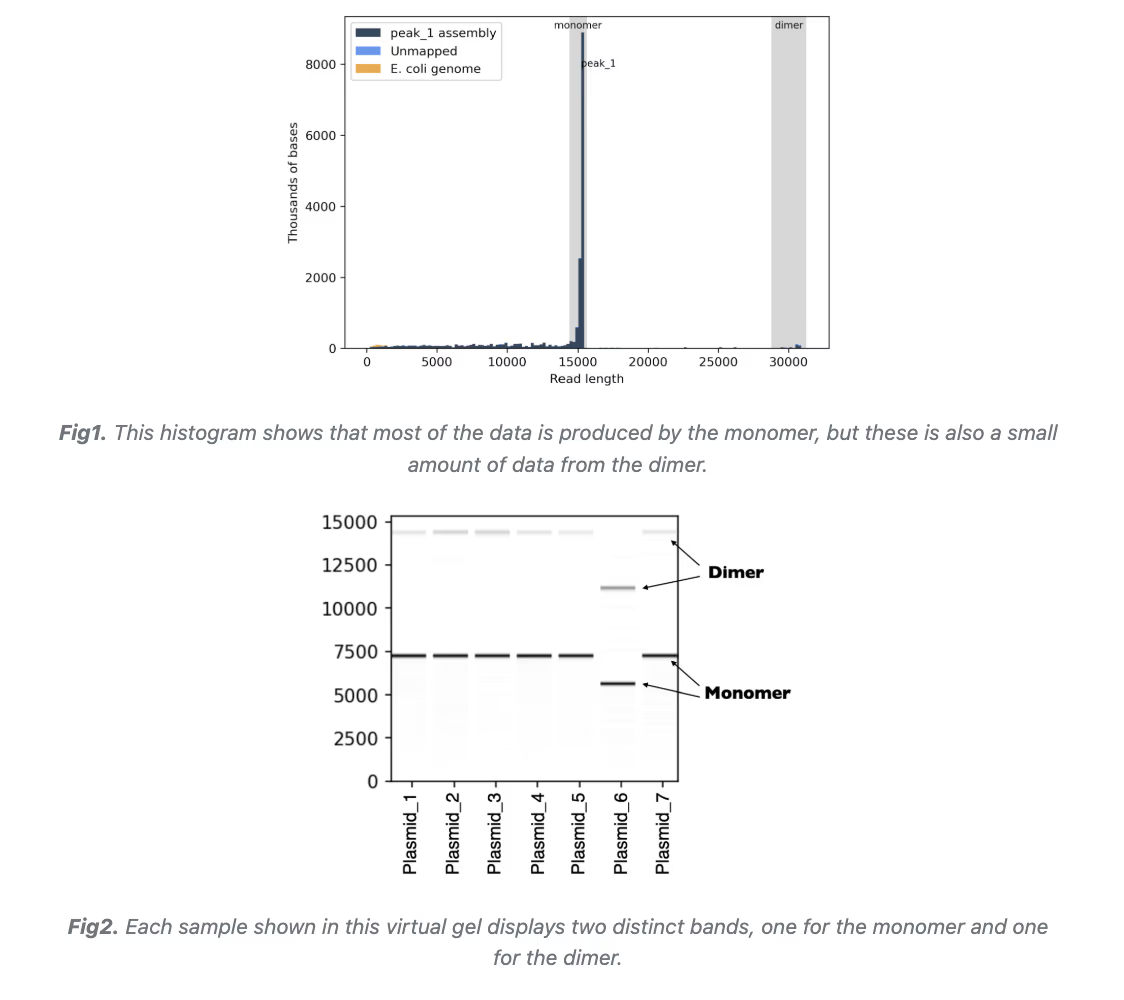
If your species are sufficiently distinct in size or sequence, the pipeline will generate a single consensus sequence for the molecular species that produces the largest amounts of total sequencing data. (Please note that multimer forms such as dimers, trimers, etc. are not considered different molecular species by the pipeline, so you will only receive the monomer consensus sequence by default).
Ultimately, which species ends up producing a consensus will vary depending on overall sample quality, coverage, and relative abundance of each species. Sequencing is considered successful if the pipeline is able to generate a consensus, even if it is not your target.
Re-sequencing mixtures won't change the relative proportions of the species (and hence which species generate a consensus), but you can submit multiple aliquots if you need higher overall coverage. Custom sequencing is available to sequence mixed populations (e.g. large barcode or variant libraries). You can submit requests for custom projects via the Custom sequencing page.
We do not guarantee a specific level of coverage as the number of raw reads generated can vary due to length and sample quality, though typically samples yield thousands of raw sequencing reads. These go through multiple quality filtering steps to ensure we are delivering the best possible assembly.
Average coverage is reported in the SAMPLE_summary.tsv file, and coverage over ~20x indicates a very accurate consensus.
Sample Handling & Preparation
There are a few ways this could be done. The option that would save you the most time would be to resuspend your colony, reserve some either on a plate or in liquid culture to regrow, and send the rest to us.
When you pick your colony, resuspend it in water in the tube you send to us and then either:
- with the same pipette tip, restreak it on a fresh plate so that it will grow again, or
- take a small amount (~1μl) of the liquid to reinnoculate into fresh media and incubate
Troubleshooting
Nanopore sequencing has two known error modes: methylation sites and homopolymers with a length >9. In both cases, in order to prevent this causing any downstream issues, our bioinformatics pipeline clearly labels these errors in light blue:
If you are interested in the details, we have more in depth explanation here.
We see concatemers like this all the time—they are not a sequencing artifact! Sanger sequencing can't detect them nor will you see them gel of your digested/linearized plasmid because the restriction sites get multiplied too. They turn out to be a lot more common than people thought. If you run your sample uncut on a gel with a supercoiled ladder, it will confirm the multimer bands.
They seem to be formed in vivo during growth in a RecA+ strain (such as NEB Turbo cells), and are more common when plasmids have large repetitive regions or other complex structures. Plasmid manufacturers like Addgene observe that concatemers occur frequently.
Please note that multimers are not considered different molecular species by the pipeline, so you will only receive the monomer consensus sequence, even if other concatemer forms produced more sequencing data.
For plasmids, "failure" means that your sample did not produce data of sufficient quality and quantity for the pipeline to generate a consensus sequence.
Our low sequencing prices and fast turnaround times do not include extensive QC to determine why your plasmid samples failed (or had low coverage). Although we do not provide definitive reasons for failure, by far the most common reasons are:
- Samples are not prepared at the required DNA concentration.
The most common cause of this is using a Nanodrop to quantify DNA concentration. We strongly recommend using a Qubit or equivalent.- You may see evidence of this failure mode in the low amount of total data reported in the raw read length histogram and in the low consensus coverage reported in the SAMPLE_summary.tsv file.
- Samples contain a mixture of plasmid species and/or fragmented genomic DNA or fragmented plasmids.
- You may see evidence of this failure mode in a wide range of read lengths reported in the raw read length histogram.
To achieve optimal sequencing results, please follow our recommended sample prep instructions.
You are welcome to submit a rerun request for any failed sample through your Order Info page or via the support@plasmidsaurus.com email address. We will evaluate whether you sample quality and quantity permits rerunning the sample (and we may also ask you to provide a reference sequence).
Sample quality checks may require any of the following:
- Quantify concentration and/ or purity
- Sample clean-up using the Bead Clean Up Protocol ($5 per sample)
- Normalizing concentration
If you have questions about your results, please get in touch at support@plasmidsaurus.com.