
How does nanopore sequencing work?
Nanopore sequencing works on the principle of measuring the change in current over time as a piece of DNA passes through a nanopore embedded in an electrically resistant membrane. Each of the four canonical DNA bases have a specific shape and size, and hence block the current by a characteristic amount. Machine learning algorithms are then able to convert the raw current “squiggle” into the base sequence.
Methylation
Modified bases, for example 5-methylcytosine (5mC) or 6-methyladenosine (6mA), also have a unique shape and size. The added methyl group shows up in the raw squiggle distinctly from a regular cytosine. E. coli plasmids have two common methylated motifs: the Dam motif Gm6ATC and the Dcm motif which can be either C5mCTGG or C5mCAGG.
Because these bases are distinct from their canonical variants, the motif containing them will be represented by an alternate current trace, which can cause the basecaller to misclassify one or more of the bases within the motif. These show up as systematic errors when you align to the reference. In fact, one early method of looking for base modifications was to look for these systematic errors.
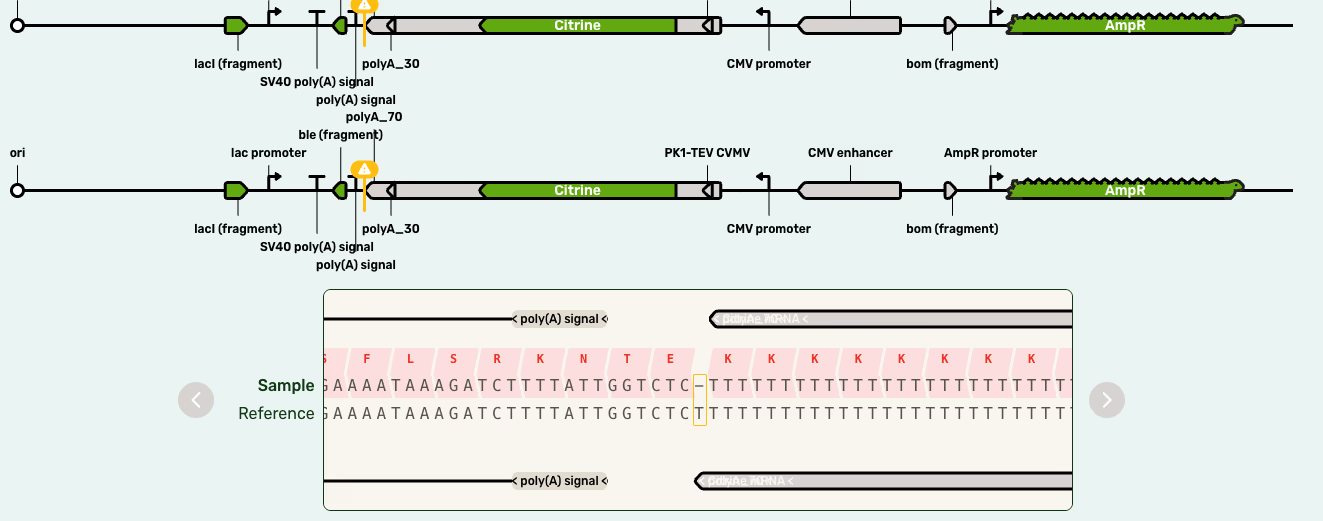
However, we have a solution. After sequencing to the appropriate depth, we assemble the most abundant plasmid sequence. This assembly is then polished using a bacterial methylation-aware algorithm that was trained on datasets containing common methylation motifs, and the vast majority of the time this fixes the error (Figure 1).
Homopolymers
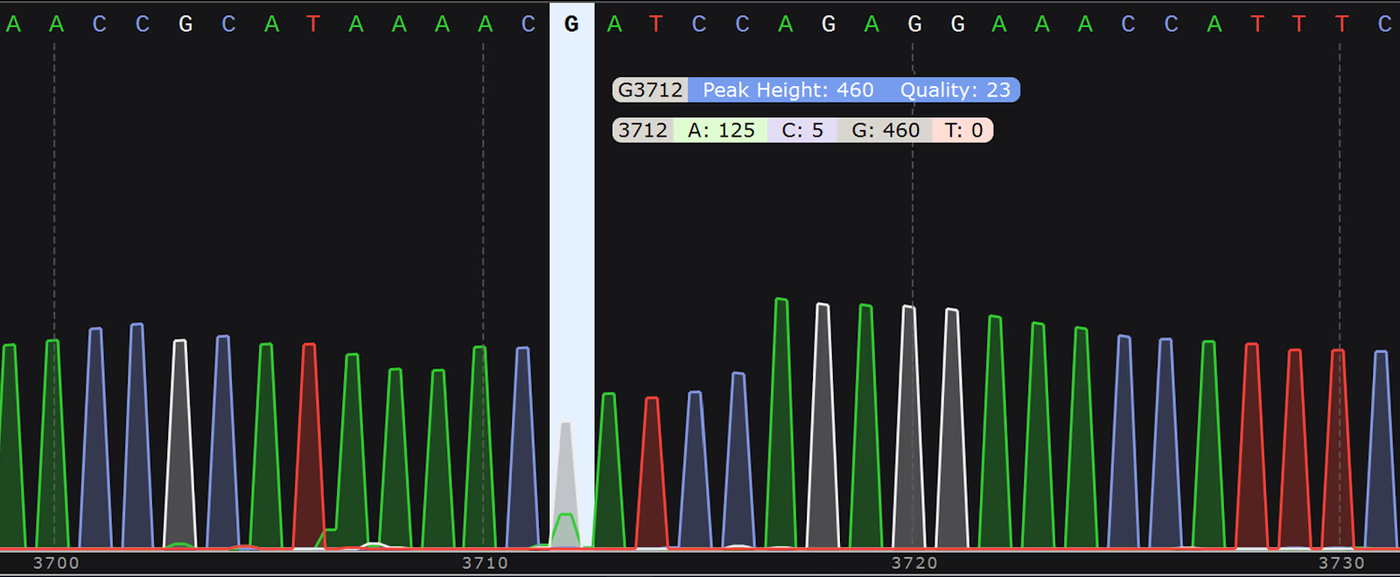
Homopolymers are stretches of DNA which contain only a single base. Their precise biological functions remain to be fully elucidated, but they appear to have regulatory functions. They pose a problem for all sequencing technologies, in Sanger-derived NGS technologies due to DNA polymerase slippage, and in nanopore sequencing because of the lack of variation.
As we saw above, nanopore sequencing works on the principle of measuring the change in current as the DNA moves through the pore. The basecalling algorithms use the interaction of the dwell time and the current level to call the base, but when the current is not changing because of the homopolymer, it becomes more difficult to accurately call the length.
The basecaller can always tell you which nucleotide makes up the homopolymer, but once the length is >9 the resulting fastq sequence is often truncated by a base or two. In most cases, simply knowing that this is likely to happen with homopolymers will allow you to correct any predicted frameshift (Figure 2).
Plasmidsaurus glyphs
Because these two types of error are well documented, our bioinformatics pipelines look for them specifically. This allows us to warn you when a mismatch between your sequencing result and your reference is more likely to be due to a sequencing error rather than a true issue with your DNA. Mismatches in the reference alignment that cannot be attributed to these common sequencing error patterns are almost certainly real and will be colored red and labelled “Mismatch”:
While mismatches that belong to either of these two categories (methylation error or homopolymer error) will be colored light blue and labeled “Likely Match”, because it is likely that your DNA actually does match the reference and the mismatch is a sequencing error rather than a true mutation:
Sample misclassification
Demultiplexing error is a well-documented phenomenon that affects NGS platforms. It occurs when a sequencing read from one sample becomes erroneously associated with a different sample. This can happen through any of a number of different mechanisms that vary in severity from one NGS platform to another.
Plasmidsaurus’s scientists and bioinformaticians are experts at preventing, detecting, and removing these improperly classified reads. Upstream, we have developed rigorous sample handling robotics and proprietary in-house chemistry which prevents ligation of the barcode to the wrong sample. Downstream, we implement rigorous bioinformatic QC steps which identify barcode misclassification, even at very low levels, and remove it before it can cause problems with results.
In addition to causing incorrect and confusing results, sample misclassification can cause sensitive intellectual property to be released to the wrong customer. We take this issue extremely seriously and we are confident that our obsessive focus on data quality and security results in the lowest sample misclassification rate of any service provider. In cases where data security is of the utmost importance, we provide the option to purchase an entire flowcell through our Custom sequencing service for maximum assurance.