Amplicon Sequencing Technical Documentation
Technical Details
Plasmidsaurus Genotyping Analysis is an amplicon sequencing service that allows you to characterize the allelic composition and frequency at specific loci. By leveraging Oxford Nanopore long-read sequencing, this service empowers you to confidently phase variants and easily sequence extended amplicons (100 bp -25 kb), providing a comprehensive view of your targets. Genotyping Analysis accurately detects all alleles present at >5% frequency.
Sample Preparation
Official sample preparation instructions are outlined on the Amplicon Sample Prep page.
Data deliverables and file types
- Data tables for allelic sequences (.tsv) | The “variants” folder reports base call differences, relative to a reference, and their frequencies, for each allele. The “allele-counts” folder contains total counts supporting each allele, displayed in tables.
- Virtual gel (.png) | Shows the distribution of read lengths in your sample - great for quickly identifying samples with unexpected PCR products.
- Allele sequence files (.ab1) | The “ab1-files” folder contains chromatograms for each allele. The “alleles” folder contains the sequence of alleles in fasta format. Allele-genbank-files provide annotated genbank files for each allele.
- Feature annotation and reference alignments | Plasmidsaurus-provided annotations are automatically provided in the feature map (online only). Automatic reference alignments also identify mismatches and associated protein consequences, between your reference and alleles, so you can immediately characterize your genetic variation.
- Sequencing statistics including read length and coverage | Histograms characterizing coverage distribution across read lengths as well as other sequencing metrics.
- Raw reads | Raw reads mapping to your sample are provided in fastq format.
Troubleshooting
PCR-mediated recombination artifacts
Recombination happens during multi-template PCR amplification when incomplete polymerase extension products from one template anneal to a different, homologous template in subsequent PCR cycles, creating chimeric (hybrid) molecules. This phenomenon, sometimes called ‘template switching,’ can compromise haplotype phasing, as artifactual recombination misrepresents the true allelic combinations on a given chromosome.
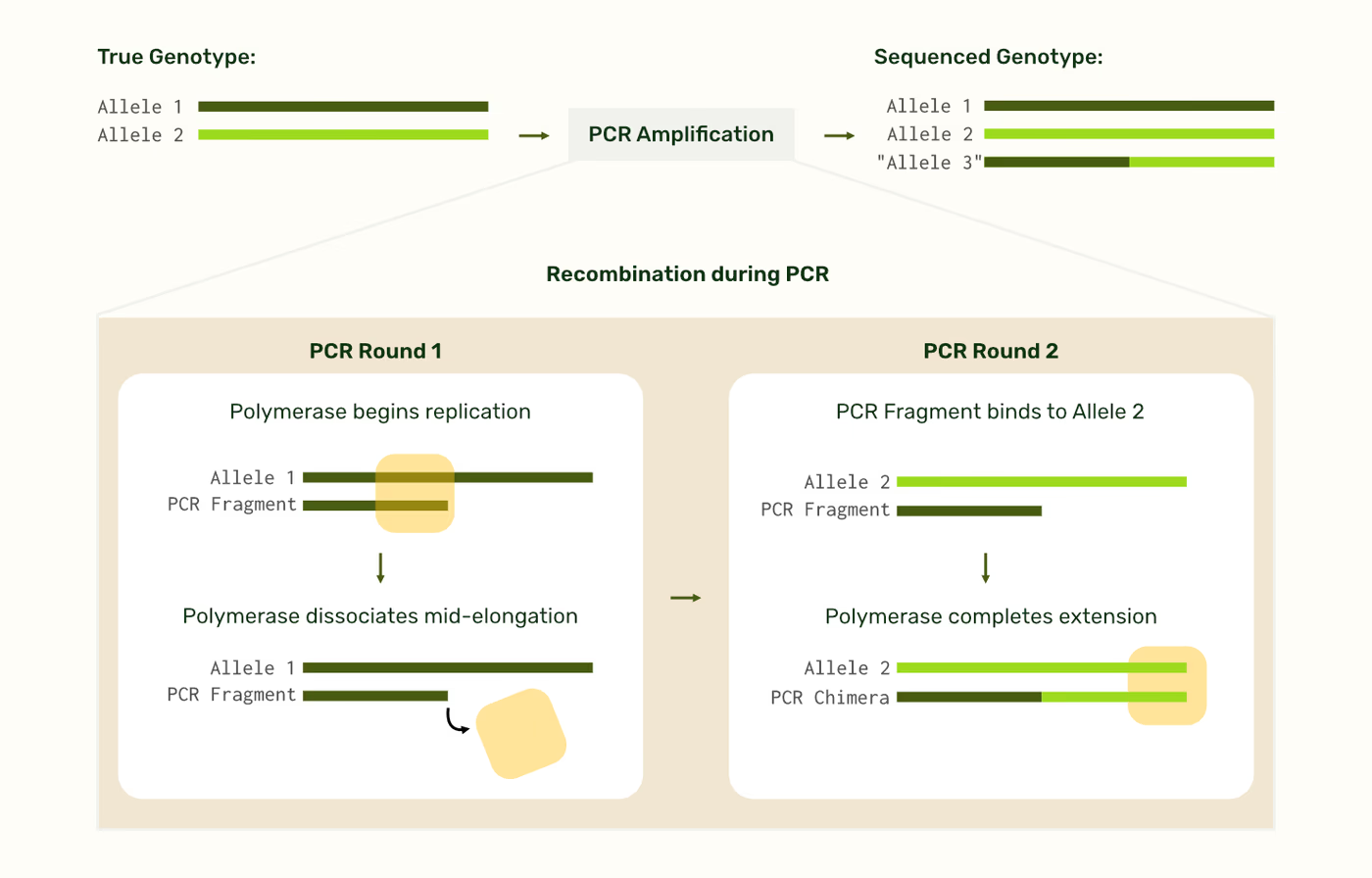
What does recombination look like?
Since there are instances where multiple alleles exist, like polyploidy, our system does not flag recombined chimeras as artifacts, it simply reports their fraction of the population like a normal allele.
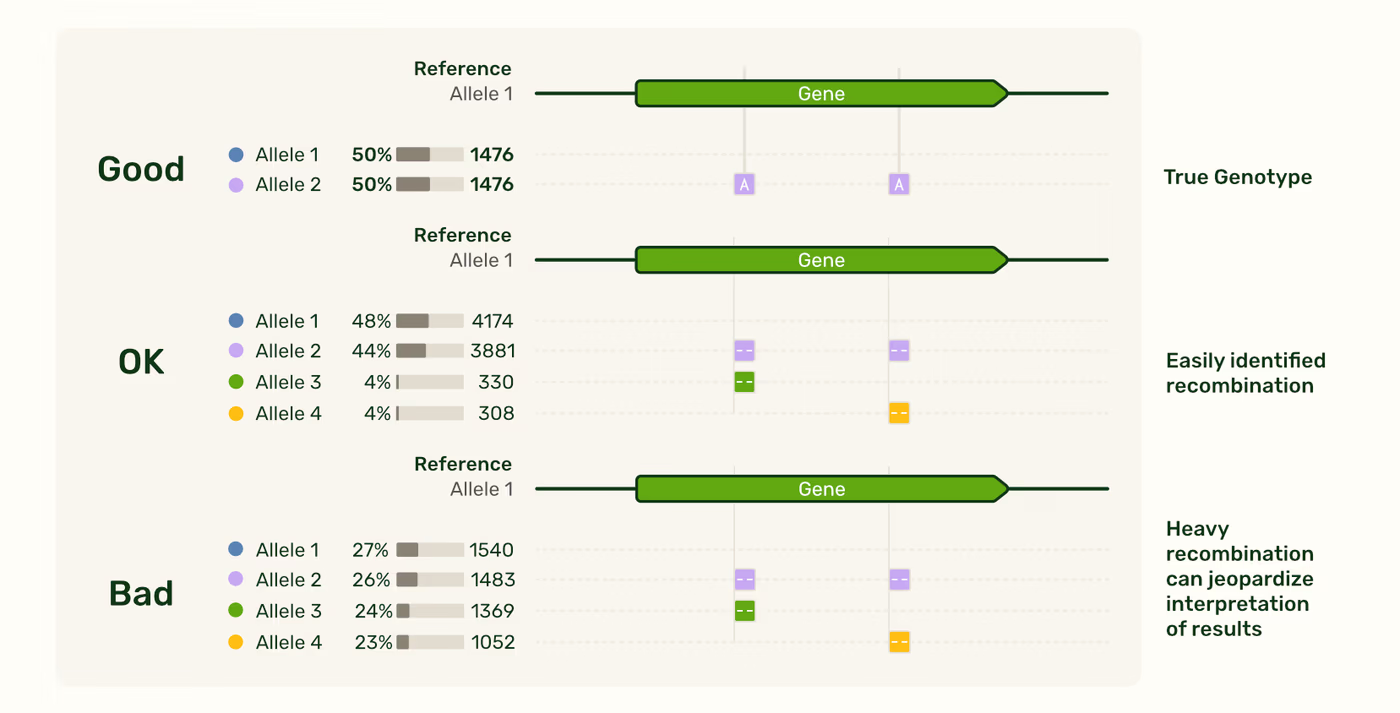
In very simple cases, it may be easy to identify this chimeric population as a low level artifact, allowing interpretation of the sample’s true genotype. For instance, when analyzing a diploid sample with two known and expected alleles, the recombined alleles might easily be ruled out as artifacts. However, in other cases, identifying recombination may not be as straightforward. In the case of polyploid analysis, ruling out chimeras by logic can be difficult. If you see evidence of chimerism in your results or are worried about how it may affect the interpretation of your results, read on to learn about approaches to mitigate this risk.
What can I do to prevent recombination?
The underlying mechanisms of PCR-mediated recombination are well-documented, providing researchers with established strategies to control its frequency during amplification.
The most important factor for minimizing recombination is to choose a polymerase with high processivity. Processivity is dictated by the intrinsic properties of the enzyme, and different polymerases possess unique template-binding mechanisms and dissociation rate constants that influence how many nucleotides are added before it falls off the template. Processivity can also be influenced by the components of the reaction such as nucleotide concentration and choice of buffer, as well as the specific composition of nucleotides on the DNA template sequence.
To help our customers find ideal PCR conditions to minimize recombination, we performed a series of optimization experiments to reduce recombination to an undetectable level in our hands.
We performed the optimization using a sample of known allelic composition, made up of a 50/50 mixture of allelic sequences, 1.6 kb in length, that differ by two mutations. We then prepared PCR amplicons varying the polymerase, template concentration, and number of PCR cycles. Then, we sequenced the amplicons and summed the percentage of recombined chimeras reported in each sample. For example, this sample contained 18% of both chimeric sequences, so the reported recombination frequency would be 36%.
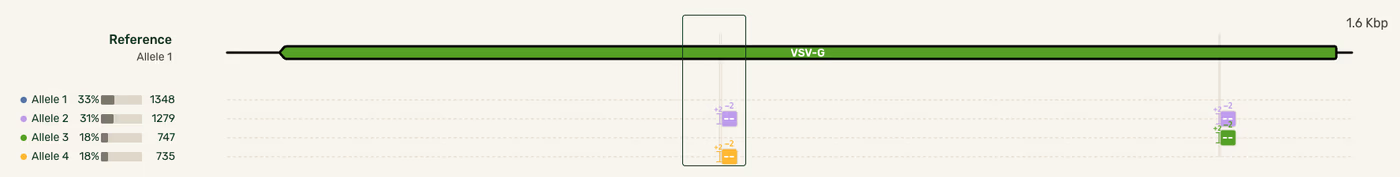
Outcomes of Plasmidsaurus’ Genotyping Analysis PCR optimization
As expected, the choice of polymerase had the biggest impact on recombination rate. We tested a few commercial polymerases (not shown) well known for high processivity, and contrasted them with a polymerase often used to deliberately recombine DNA, Taq. We found that the Takara Primestar GXL Polymerase was best for minimizing recombination in our hands.
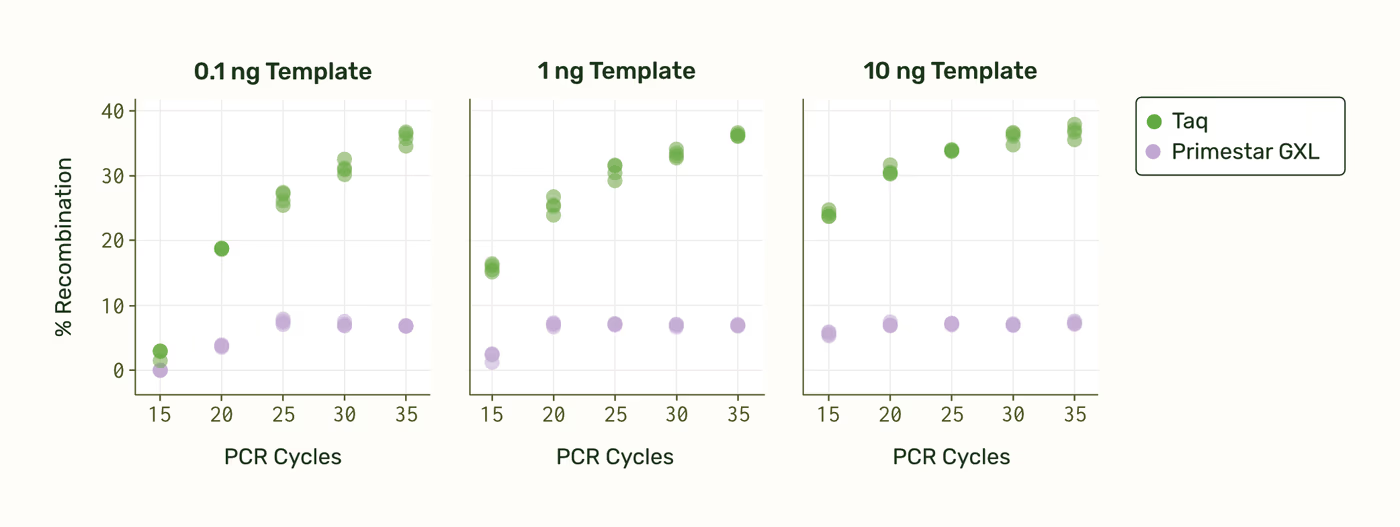
We also found starting template concentration and number of PCR cycles to be important factors. We monitored Primestar GXL reaction progress using qPCR, and sequenced samples removed at 15, 25, 30, and 35 cycles.
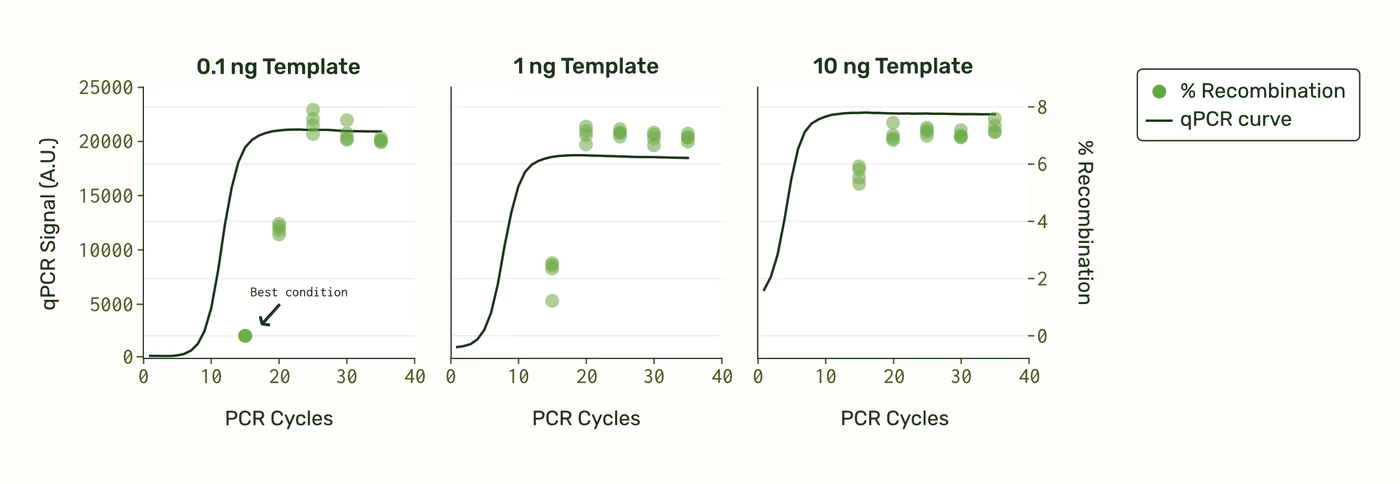
Recombination appears to lag behind the qPCR sigmoid curve. This is perhaps because as the reaction progresses and dNTPs are depleted, polymerase stalling and template dissociation occur more frequently, leading to more PCR fragments. We noticed that the best condition that led to undetectable recombination falls before the qPCR sigmoid curve inflection point, while dNTPs and primers are still abundant. However, once all resources have been fully consumed, the rate of recombination stays constant for the remainder of the cycles (because amplification has ended).
The conditions that led to undetectable recombination for the 1.6 kb amplicon were:
- 0.1 ng template
- Primestar GXL Premix (2X master mix)
- Primestar GXL protocol for ≤10 kb products:
- 98°C 10 sec
- 55°C 15 sec
- 68°C 1 min/kb
- Repeat 15 cycles
Conclusion and recommendations
We recommend using a polymerase with high processivity as a starting point for your amplicon preparation PCRs. While reducing the DNA template and PCR cycles is generally useful, you must carefully balance the recombination rate with total yield to ensure sufficient material for robust sequencing. Because PCR performance can vary based on template length, template quality, primer design, and thermal cycler used, you may need to perform optimization to find conditions that most effectively reduce recombination for your specific amplicon.
Literature suggests performing optimization with a control sequence, such as the one we performed, is the best way to minimize artifacts and obtain reliable results from multi-template PCRs. We found the qPCR-based experiment to be the simplest and most informative way to find conditions that maximize yield and minimize recombination.
Amplicon size bias
What is amplicon size bias?
Amplicon size bias is a phenomenon where shorter DNA fragments are preferentially amplified during PCR, resulting in an underrepresentation of longer fragments and skewed amplicon abundance. This is a common problem in multi-template PCRs; even small differences in amplification efficiency between templates can lead to vastly different template ratios after exponential amplification. This bias can depend on the relative difference between amplicon lengths as well as the overall amplicon size.
For genotyping samples with large differences in amplicon length (such as insertions or deletions), the reported allele frequencies may not reflect the true population. If quantitative results are necessary, contact Plasmidsaurus' Custom Sequencing services.
Rerun Policy
You are welcome to submit a rerun request for any failed sample through your Order Info page or via the support@plasmidsaurus.com email address. We will evaluate whether your sample quality and quantity permits rerunning the sample (and we may also ask you to provide a reference sequence).
Sample quality checks may require any of the following:
- Quantify concentration and/ or purity
- Sample clean-up using the Bead Clean Up Protocol ($5 per sample)
- Normalizing concentration
If you have questions about your results, please get in touch at support@plasmidsaurus.com